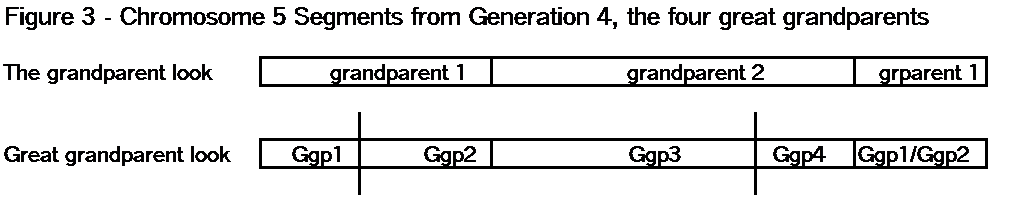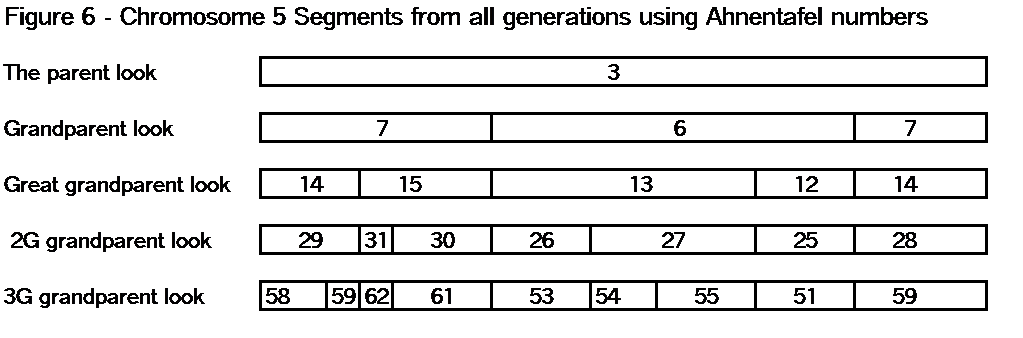The data we get from our atDNA test is not as precise as it may appear to be. But don’t worry, it doesn’t make any difference…
Let’s start with the 3 ways we measure our DNA first (see also my blog post Measuring Segments).
Base Pairs: Our DNA has about 3.2 billion base pairs in each set of chromosomes. The Human Genome Project sequenced about 99 percent our chromosomes in 2003. Since then, scientists have continued to refine the structure and arrangement of the base pairs. Most of our atDNA tests have been based on “Build 36”, but FTDNA shifted to “Build 37” a year or so ago. Because the atDNA tests only look at about 700,000 of those base pairs, the change from Build 36 to 37 is not much different. But most of us “lost” a few Matches and “gained” a few Matches as a result. To my knowledge, 23andMe and AncestryDNA still use Build 36, as does GEDmatch. The differences are slight.
cMs: The cM is an empirical measurement. There are differences in the observed cMs for males and females over the same segment (same start and end location). An average is used because the companies just don’t know the male/female ancestry of each shared segment. Besides, an average is much easier to work with… Even the tables of averages differs by company. Here are the totals per the ISOGG/wiki (without Chr X) [1]:
23andMe 3,537 cMs
FTDNA 3,384 cMs
GEDmatch 3,587 cMs
Per the CRC Research Group [2] we have very different totals of the average for males and females in the 22 autosomes:
Male 2,809 cMs
Female 4,782 cMs
Average 3,795 cMs
The reason this last average is greater that the averages used by the three companies is because there are certain areas of certain chromosomes that are blocked out from atDNA testing for genealogy (see the greyed area in your chromosome browser). So the companies only take the average of the areas covered (sampled) by the SNPs.
The important point to understand is that there is a wide variation between males and females. Using an average pretty much guarantees some inaccuracy. So definitely use cMs as a guideline, but don’t split hairs or make hard and fast “rules” with the cM values we get from the testing companies
SNPs: Each of the atDNA companies use a different number of SNPs per the ISOGG/wiki Comparison Chart [3]:
23andMe 577,382 SNPs
FTDNA 708,092 SNPs
AncestryDNA 682,549 SNPs
Note that 23andMe now uses a somewhat lower number of SNPs, and some of them are different from the other companies. But since the SNPs are basically a sampling technique over all of our DNA, we see little differences in the shared segments now reported by 23andMe.
So the start and end location of shared segments will tend to be different depending on where the terminal SNPs are. You might think with the largest number of SNPs, FTDNA can report a more accurate shared segment, but read on…
Shared segments are determined by a proprietary algorithm at each company. I don’t know them. And even if I did, I shouldn’t report it.
When determining 700,000 or so SNPs it’s hard to get all of them read correctly. There are invariably a few miss-calls and no-calls. Each company’s algorithm has an instruction as to handle these: ignore one (or a few?) of them and report a longer shared segment; or let a miss-call or no-call break up an otherwise long segment. Based on GEDmatch examples of kits from 2 companies for the same person, each company probably does it differently.
There is no “signpost” in our DNA to indicate where an ancestral segment starts or ends. Each algorithm looks for matches between two kits, and it may well run beyond the boundaries for a particular ancestor, and in these cases pick up, random, but matching, pieces of DNA from other ancestors, which would not be IBD. This makes some shared segments look larger than they should be. A classic example is a parent-child-Match trio where the child-Match shared segment is a little larger than the parent-Match shared segment.
The algorithms may also include some shortcuts. Notice the large number of segments at FTDNA which end in “00”. It appears some of their algorithm is based on looking at blocks of 100 SNPs at a time – in this case the size of a shared segment would be rounded down to only the blocks of 100 that match. The advantage FTDNA might have because of more SNPs in total, is offset by using them in blocks.
Here is a quote from the 23andMe Family Inheritance: Advanced page: “… segments can be measured in centiMorgans or in base pairs for mapping onto the genome. 23andMe rounds the segment length to the nearest tenth of centiMorgan and segment start and end coordinates to the closest millionth base pair to reflect the uncertainty in the exact locations of the segment boundaries.” [bolding by me]
AncestryDNA states their algorithm culls out segments based on “population” phasing. They also eliminate some pile-up segments, although it’s not clear (to me anyway) what size range they consider for this culling process. This process eliminates some IBS segments, but is also eliminates some IBD segments, too.
All of the above factors, and probably more, result in the DNA data being a little different, depending on the company – we can say the data is a little fuzzy, and not quite as precise as it appears to be. With this knowledge, I’m pretty sure cMs values are not accurate to two decimal places, and shared segments are not precise to a particular base pair.
Clearly this fuzzy data leads to fuzzy segments. The statement by 23andMe is a good one – round the segment start and end locations to the nearest million base pairs.
So let’s look at all of this fuzzinessfrom the Big Picture of Chromosome Mapping with Triangulated Groups – is it significant?
Each TG is a collection of overlapping shared segments (which match each other). As far as defining a TG is concerned, the only two SNPs that count are the first one and the last one. These SNPs are often from one of the segments that overran a little – either at the start or the end location. So the TG may be a little larger than indicated – the ends of the shared segments are a little fuzzy, so the TG is a little fuzzy, too.
When Chromosome Mapping is complete, you should have a bunch of TGs that cover each chromosome, from one end to the other (see Segments: Bottom-Up). I often use chromosome 5 in examples because it is about 200cM, and averages about 2 crossover points per generation. In my Chromosome Map, so far, I have 9 paternal TGs and 11 maternal TGs that cover my two chromosome 5s. From the detailed data, the tips of these TGs may overlap a little. By a little I mean maybe up to 1 or even 2 Mbp. Don’t let it worry you. In the Big Picture, if you have 10 TGs with various Common Ancestors on chromosome 5, you’re doing great! You’ve won! The fact that you don’t know precisely where each shared segment or TG starts or ends, pales when you run Kitty’s Chromosome Mapper [4] and see your ancestors mapped across chromosome 5. The little fuzziness is lost in the Big Picture. When you finish a jigsaw puzzle and step back to admire the full picture, you don’t even notice the outlines of each puzzle piece. For genealogy, you have achieved your objective – notwithstanding the fuzziness.
Another clue that fuzzy data and fuzzy segments are not an issue, is that my Matches who have tested at multiple companies, still share virtually the same segments with me. The shared segments are almost never identical in start/end locations, cMs or SNP counts. However, they always show up within a few rows of each other in my sorted spreadsheet. And they always wind up in the same TG!
So if you want to make life (TGs and Mapping) easier, use Mbp for segments. And since they are just guidelines, round off cMs to the nearest whole number. It won’t hurt anything. For me, overlapping segments and cM thresholds are just guidelines to group segments, and then form TGs (see How To Triangulate)
And if, someday, you decide to tackle genes, and you need to know exactly which ancestor gave you the gene that appears to straddle two TGs (meaning two ancestors), you can reexamine that junction more carefully. You can always refine the crossover points later, if you want. But for now, spend your energies in forming the TGs and determining Common Ancestors for them! I’m curious about which ancestors gave me which genes, but I doubt I’ll ever go back once my chromosomes are properly mapped to distant ancestors – I’ll be pooped and ready to try something else…
Reference links:
[1] http://www.isogg.org/wiki/CentiMorgan
[2] http://europepmc.org/backend/ptpmcrender.fcgi?accid=PMC52322&blobtype=pdf
[3] http://www.isogg.org/wiki/Autosomal_DNA_testing_comparison_chart
[4] http://kittymunson.com/dna/ChromosomeMapper.php
03A Segmentology: Fuzzy Data, Fuzzy Segments – No Worry by Jim Bartlett 20150529